{kind=link}
Single-cell analysis of upper airway cells reveals host-viral dynamics in influenza infected adults
 Biology
Biology
 Genomics
Genomics
 Immunology
Immunology
 Infectious Disease
Infectious Disease
 Microbiology
Microbiology
 Alex K. Shalek
Alex K. Shalek
 José Ordovas-Montañes
José Ordovas-Montañes
bioRxiv
April, 2020
Abstract
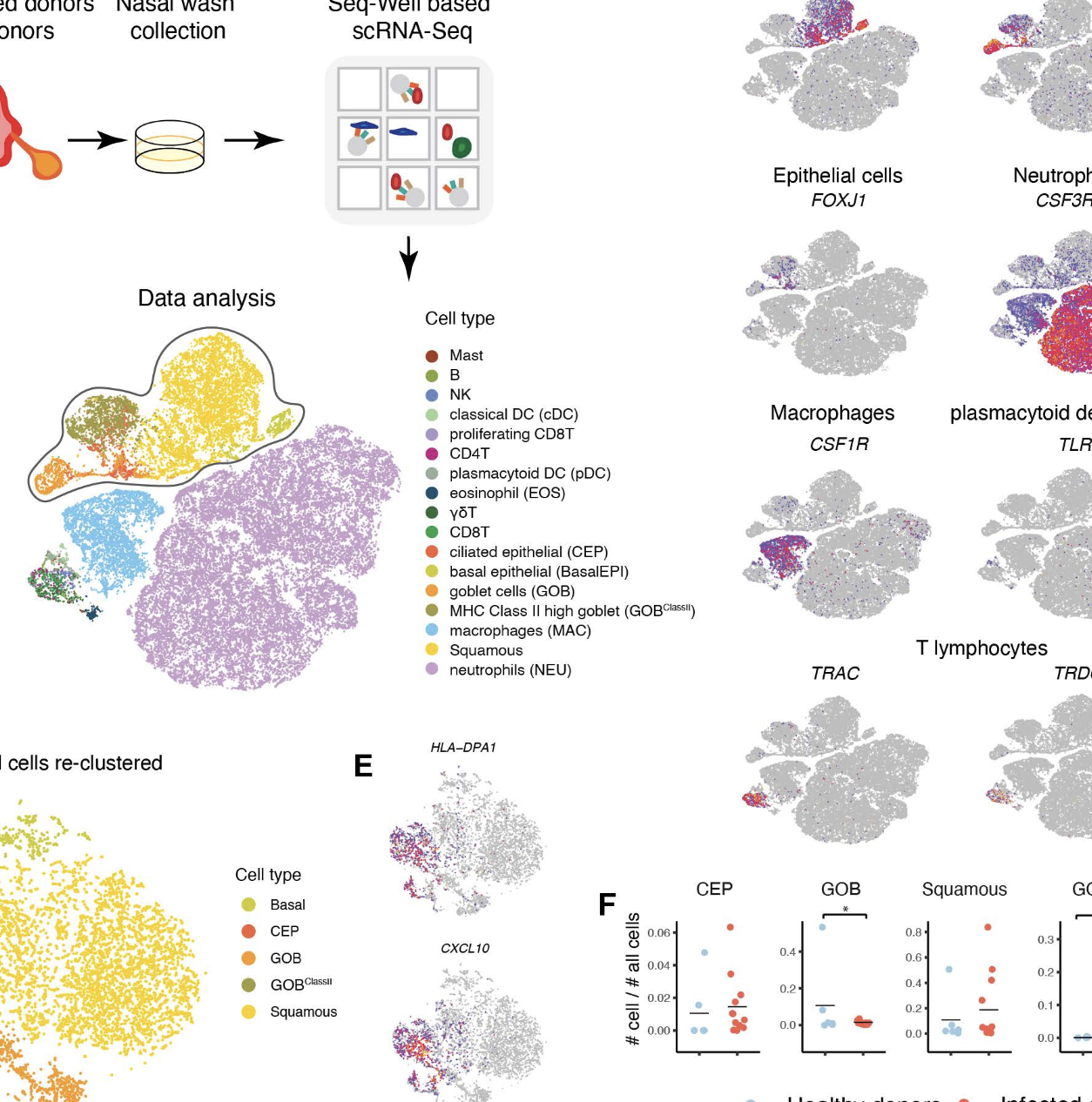
Influenza virus infections are major causes of morbidity and mortality. Research using cultured cells, bulk tissue, and animal models cannot fully capture human disease dynamics. Many aspects of virus-host interactions in a natural setting remain unclear, including the specific cell types that are infected and how they and neighboring bystander cells contribute to the overall antiviral response. To address these questions, we performed single-cell RNA sequencing (scRNA-Seq) on cells from freshly collected nasal washes from healthy human donors and donors diagnosed with acute influenza during the 2017-18 season. We describe a previously uncharacterized goblet cell population, specific to infected individuals, with high expression of MHC class II genes. Furthermore, leveraging scRNA-Seq reads, we obtained deep viral genome coverage and developed a model to rigorously identify infected cells that detected influenza infection in all epithelial cell types and even some immune cells. Our data revealed that each donor was infected by a unique influenza variant and that each variant was separated by at least one unique non-synonymous difference. Our results demonstrate the power of massively-parallel scRNA-Seq to study viral variation, as well as host and viral transcriptional activity during human infection.