{kind=link}
Comparative analysis of cell–cell communication at single-cell resolution
 Computational Methods
Computational Methods
 Genomics
Genomics
 R&D
R&D
 Statistics
Statistics
 Alex K. Shalek
Alex K. Shalek
Nature Biotechnology
May, 2023
Abstract
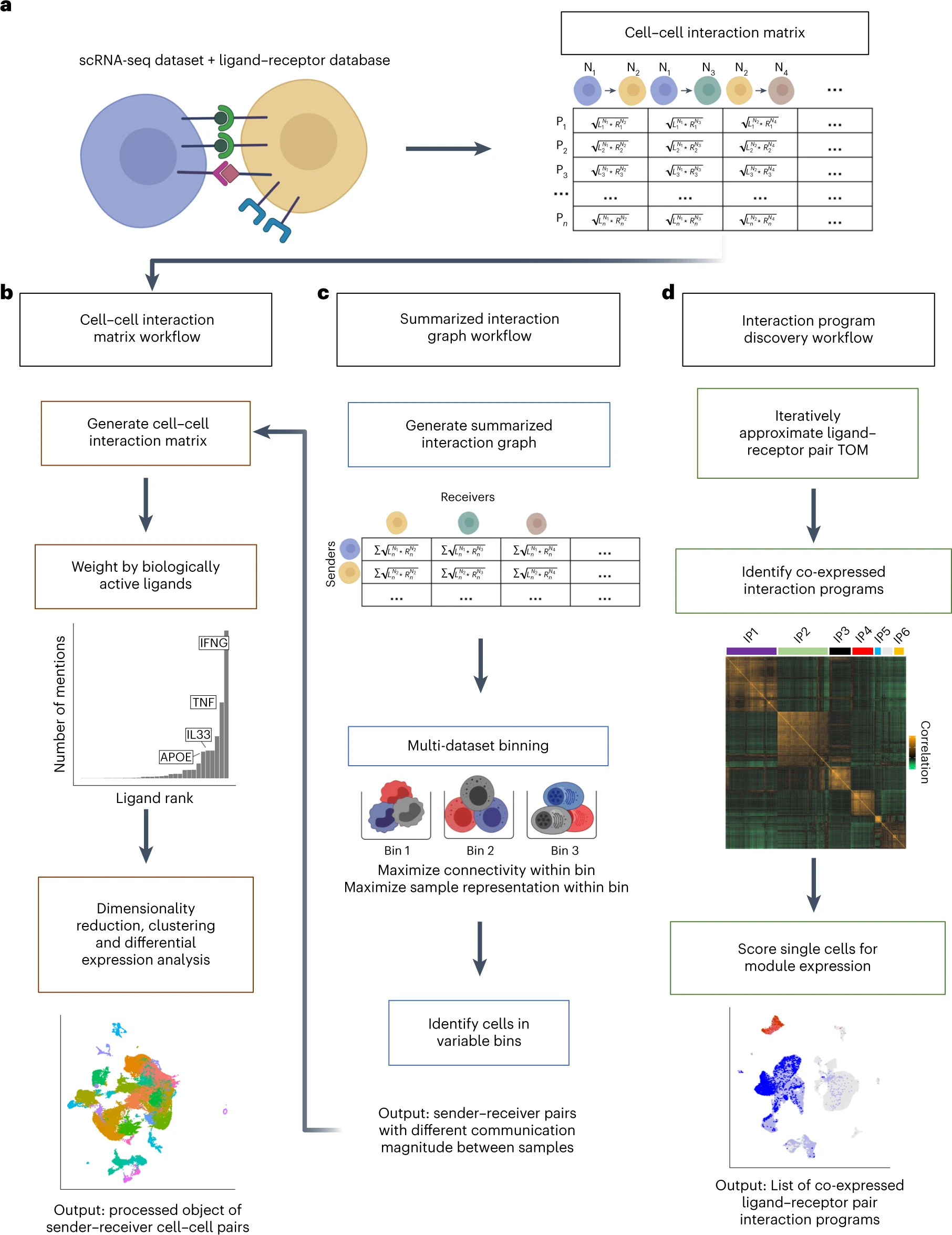
Inference of cell–cell communication from single-cell RNA sequencing data is a powerful technique to uncover intercellular communication pathways, yet existing methods perform this analysis at the level of the cell type or cluster, discarding single-cell-level information. Here we present Scriabin, a flexible and scalable framework for comparative analysis of cell–cell communication at single-cell resolution that is performed without cell aggregation or downsampling. We use multiple published atlas-scale datasets, genetic perturbation screens and direct experimental validation to show that Scriabin accurately recovers expected cell–cell communication edges and identifies communication networks that can be obscured by agglomerative methods. Additionally, we use spatial transcriptomic data to show that Scriabin can uncover spatial features of interaction from dissociated data alone. Finally, we demonstrate applications to longitudinal datasets to follow communication pathways operating between timepoints. Our approach represents a broadly applicable strategy to reveal the full structure of niche–phenotype relationships in health and disease.