{kind=link}
Compressed phenotypic screens for complex multicellular models and high-content assays
 Biology
Biology
 Cancer
Cancer
 Chemistry
Chemistry
 Computational Methods
Computational Methods
 Genomics
Genomics
 R&D
R&D
 Statistics
Statistics
 Technology
Technology
 Alex K. Shalek
Alex K. Shalek
 Ben Mead
Ben Mead
 Conner Kummerlowe
Conner Kummerlowe
 Ivy Liu
Ivy Liu
 Peter Winter
Peter Winter
 Walaa Kattan
Walaa Kattan
bioRxiv
March, 2023
Abstract
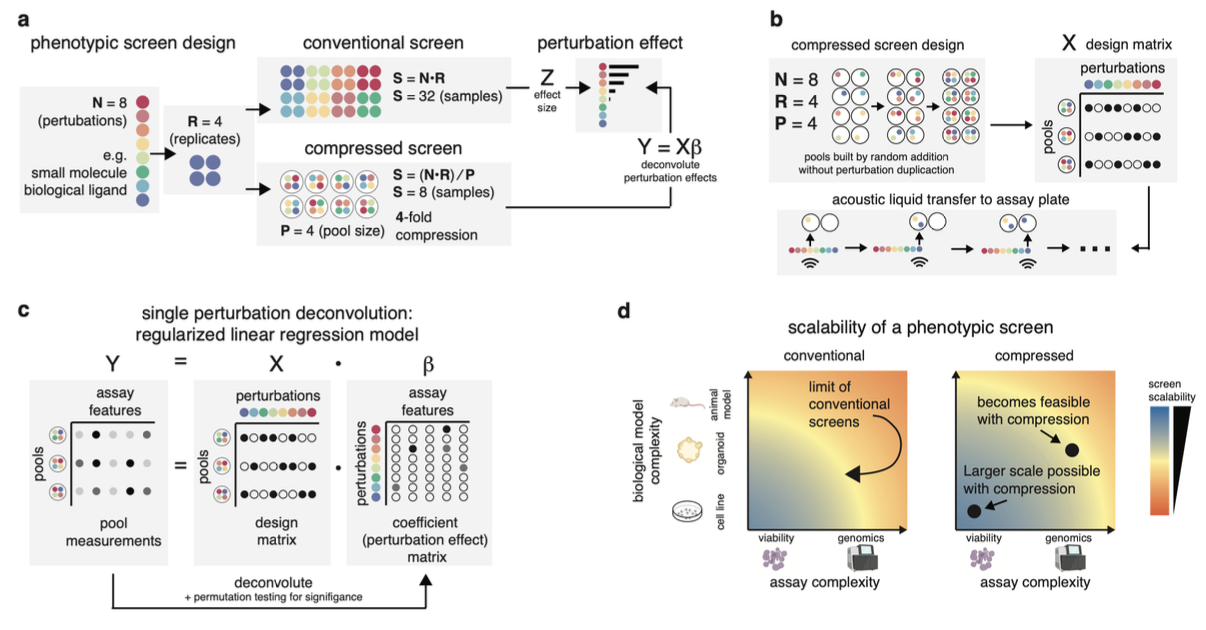
High-throughput phenotypic screens leveraging biochemical perturbations, high-content readouts, and complex multicellular models could advance therapeutic discovery yet remain constrained by limitations of scale. To address this, we establish a method for compressing screens by pooling perturbations followed by computational deconvolution. Conducting controlled benchmarks with a highly bioactive small molecule library and a high-content imaging readout, we demonstrate increased efficiency for compressed experimental designs compared to conventional approaches. To prove generalizability, we apply compressed screening to examine transcriptional responses of patient-derived pancreatic cancer organoids to a library of tumor-microenvironment (TME)-nominated recombinant protein ligands. Using single-cell RNA-seq as a readout, we uncover reproducible phenotypic shifts induced by ligands that correlate with clinical features in larger datasets and are distinct from reference signatures available in public databases. In sum, our approach enables phenotypic screens that interrogate complex multicellular models with rich phenotypic readouts to advance translatable drug discovery as well as basic biology.