{kind=link}
High-fat diet-activated fatty acid oxidation mediates intestinal stemness and tumorigenicity
 Biology
Biology
 Genomics
Genomics
 Alex K. Shalek
Alex K. Shalek
 Constantine Tzouanas
Constantine Tzouanas
Cell Reports , Volume 35
June, 2021
Abstract
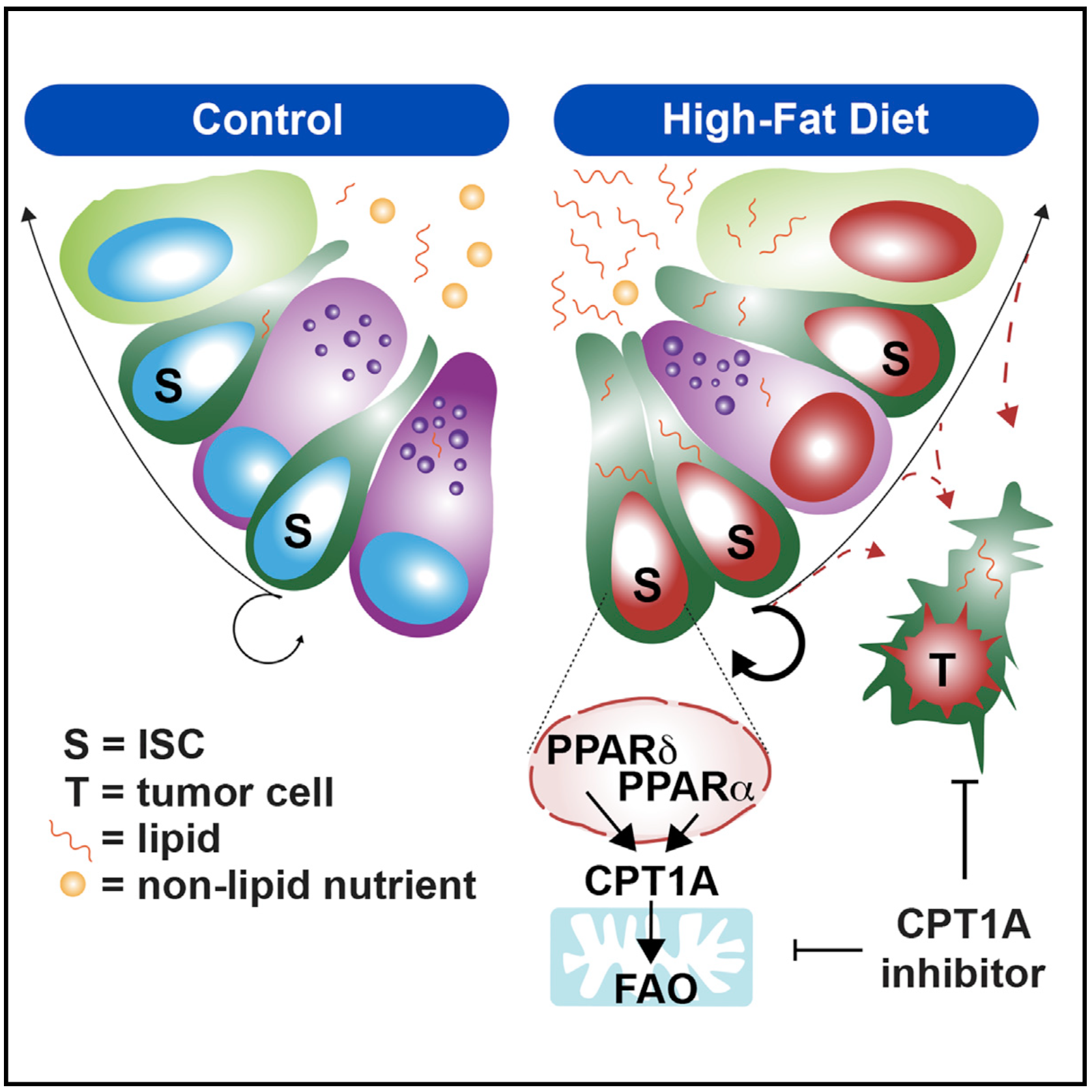
Obesity is an established risk factor for cancer in many tissues. In the mammalian intestine, a pro-obesity high-fat diet (HFD) promotes regeneration and tumorigenesis by enhancing intestinal stem cell (ISC) numbers, proliferation, and function. Although PPAR (peroxisome proliferator-activated receptor) nuclear receptor activity has been proposed to facilitate these effects, their exact role is unclear. Here we find that, in loss-of-function in vivo models, PPARα and PPARδ contribute to the HFD response in ISCs. Mechanistically, both PPARs do so by robustly inducing a downstream fatty acid oxidation (FAO) metabolic program. Pharmacologic and genetic disruption of CPT1A (the rate-controlling enzyme of mitochondrial FAO) blunts the HFD phenotype in ISCs. Furthermore, inhibition of CPT1A dampens the pro-tumorigenic consequences of a HFD on early tumor incidence and progression. These findings demonstrate that inhibition of a HFD-activated FAO program creates a therapeutic opportunity to counter the effects of a HFD on ISCs and intestinal tumorigenesis.