{kind=link}
Reduced cellular diversity and an altered basal progenitor cell state inform epithelial barrier dysfunction in human type 2 immunity
 Cell Atlas
Cell Atlas
 Immunology
Immunology
 Medicine
Medicine
 Alex K. Shalek
Alex K. Shalek
 José Ordovas-Montañes
José Ordovas-Montañes
 Marc Wadsworth II
Marc Wadsworth II
 Sam Kazer
Sam Kazer
 Sarah Nyquist
Sarah Nyquist
 Travis Hughes
Travis Hughes
BioRxiv
November, 2017
Abstract
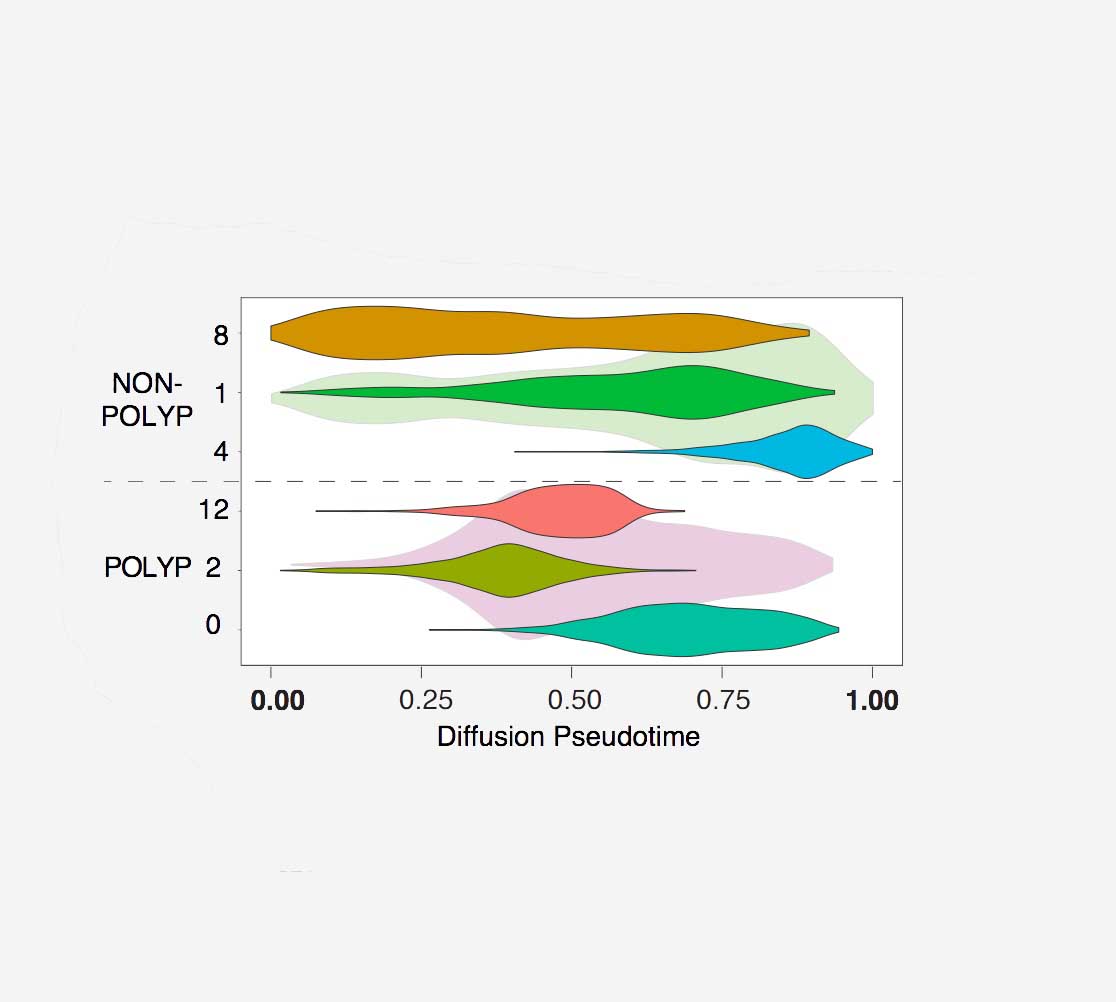
Tissue barrier dysfunction is a poorly defined feature hypothesized to drive chronic human inflammatory disease. The epithelium of the upper respiratory tract represents one such barrier, responsible for separating inhaled agents, such as pathogens and allergens, from the underlying submucosa. Specialized epithelial subsets-including secretory, glandular, and ciliated cells-differentiate from basal progenitors to collectively realize this role. Allergic inflammation in the upper airway barrier can develop from persistent activation of Type 2 immunity (T2I), resulting in the disease spectrum known as chronic rhinosinusitis (CRS), ranging from rhinitis to severe nasal polyps. Whether recently identified epithelial progenitor subsets, and their differentiation trajectory, contribute to the clinical presentation and barrier dysfunction in T2I-mediated disease in humans remains unexplored. Profiling twelve primary human CRS samples spanning the range of clinical severity with the Seq-Well platform for massively-parallel single-cell RNA-sequencing (scRNA-seq), we report the first single-cell transcriptomes for human respiratory epithelial cell subsets, immune cells, and parenchymal cells (18,036 total cells) from a T2I inflammatory disease, and map key mediators. We find striking differences between non-polyp and polyp tissues within the epithelial compartments of human T2I cellular ecosystems. More specifically, across 10,383 epithelial cells, we identify a global reduction in epithelial diversity in polyps characterized by basal cell hyperplasia, a concomitant decrease in glandular and ciliated cells, and phenotypic shifts in secretory cell function. We validate these findings through flow cytometry, histology, and bulk tissue RNA-seq of an independent cohort. Furthermore, we detect an aberrant basal progenitor differentiation trajectory in polyps, and uncover cell-intrinsic and extrinsic factors that may lock polyp basal cells into an uncommitted state. Overall, our data define severe T2I barrier dysfunction as a reduction in epithelial diversity, characterized by profound functional shifts stemming from basal cell defects, and nominate a cellular mechanism for the persistence and chronicity of severe human respiratory disease.
Click here to read the pre-publication manuscript.