{kind=link}
Systematic comparison of single-cell and single-nucleus RNA-sequencing methods
 Computational Methods
Computational Methods
 Genomics
Genomics
 R&D
R&D
 Technology
Technology
 Alex K. Shalek
Alex K. Shalek
 Marc Wadsworth II
Marc Wadsworth II
 Shaina Carroll
Shaina Carroll
 Travis Hughes
Travis Hughes
Nature Biotechnology , Volume 38
April, 2020
Abstract
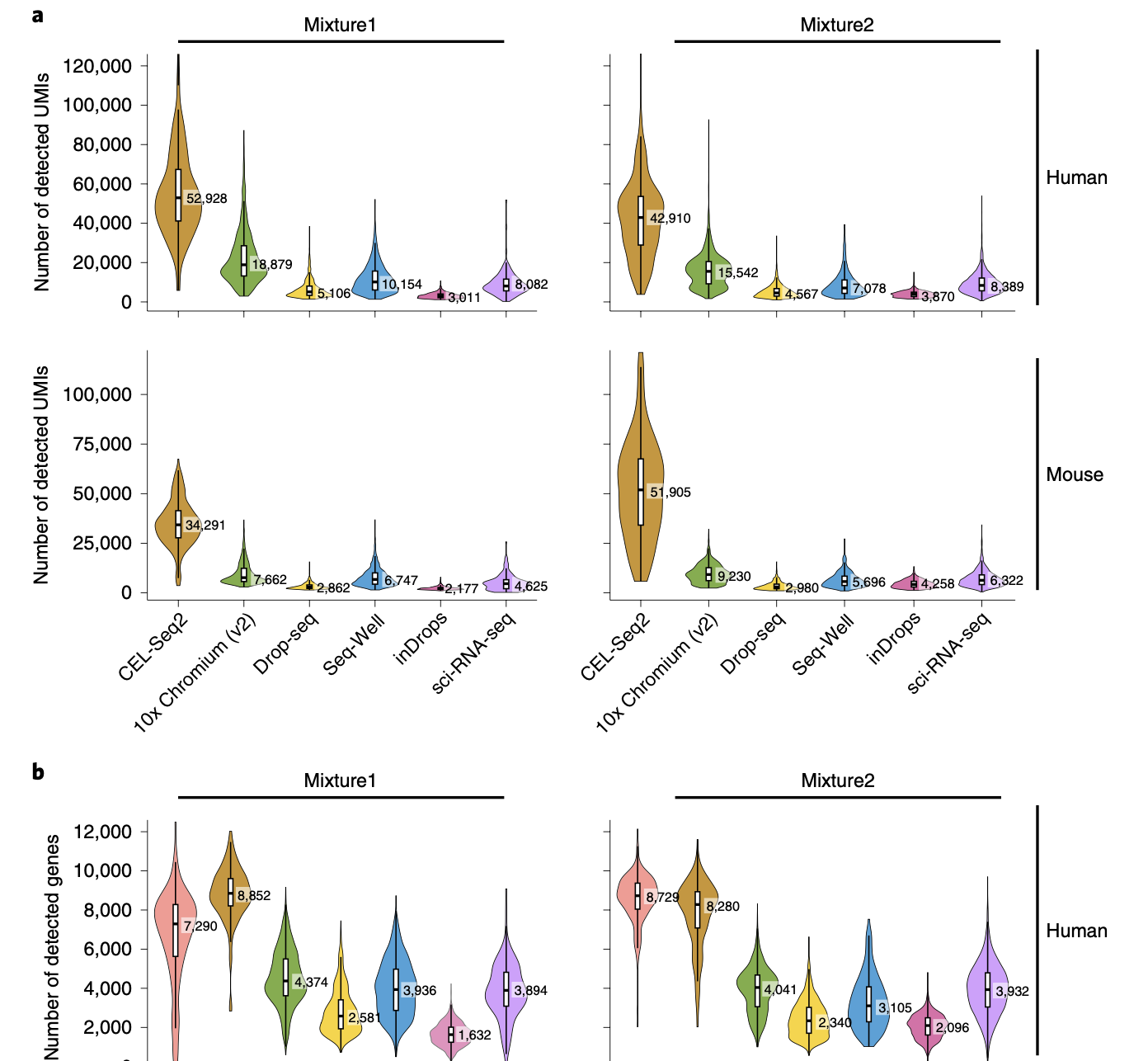
The scale and capabilities of single-cell RNA-sequencing methods have expanded rapidly in recent years, enabling major dis- coveries and large-scale cell mapping efforts. However, these methods have not been systematically and comprehensively benchmarked. Here, we directly compare seven methods for single-cell and/or single-nucleus profiling—selecting representa- tive methods based on their usage and our expertise and resources to prepare libraries—including two low-throughput and five high-throughput methods. We tested the methods on three types of samples: cell lines, peripheral blood mononuclear cells and brain tissue, generating 36 libraries in six separate experiments in a single center. To directly compare the methods and avoid processing differences introduced by the existing pipelines, we developed scumi, a flexible computational pipeline that can be used with any single-cell RNA-sequencing method. We evaluated the methods for both basic performance, such as the structure and alignment of reads, sensitivity and extent of multiplets, and for their ability to recover known biological information in the samples.