We combine genomics, chemical biology, and nanotechnology to construct accessible and widely-useful cross-disciplinary platforms that enable us and others to profile and control cells and their interactions. By applying these approaches with partners around the world, we can identify and test how cellular composition and communication drive successful or aberrant ensemble immune responses and influence homeostatic stability to environmental perturbations across organs and diseases. We are committed to sharing and training others in these technologies, both to empower global inquiry and to discover limitations to overcome.
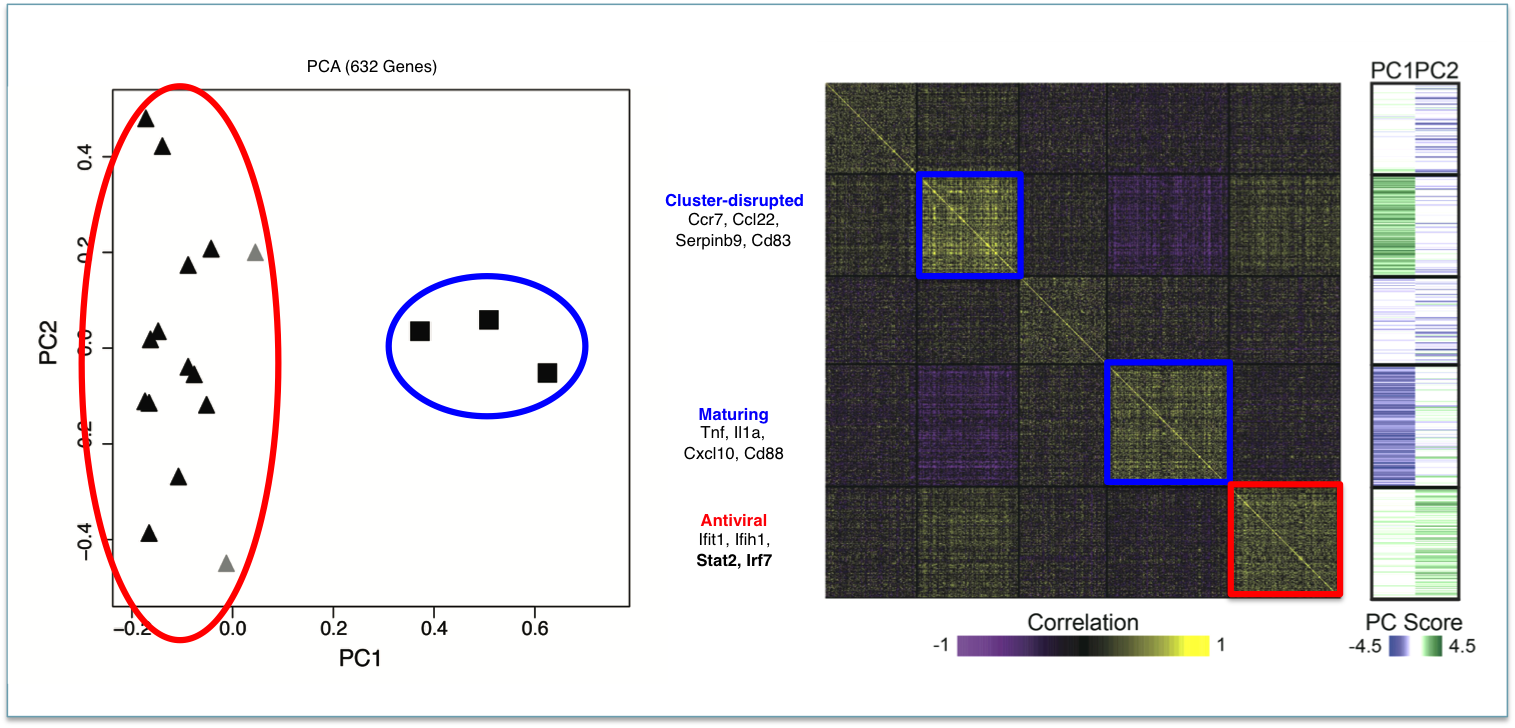
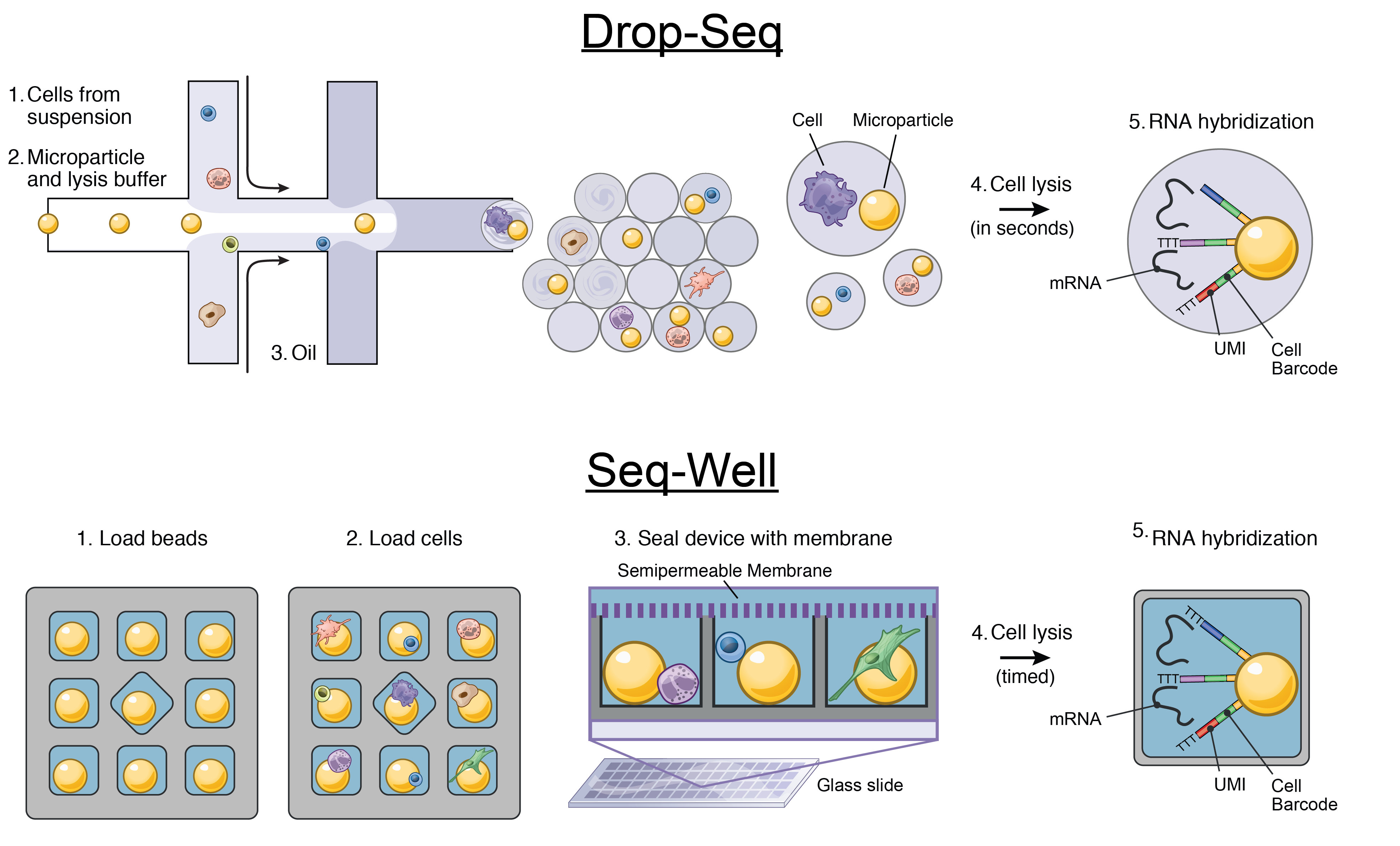
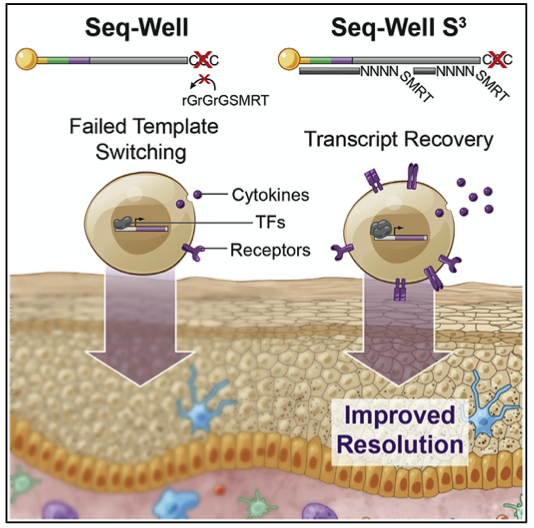
We develop single-cell transcriptomic approaches to comprehensively profile human tissues and model systems. Previously, we focused on establishing, validating, scaling, and simplifying single-cell RNA-seq, often through the development of microdevices, to enable genome-wide identification of the cell types/states contained within complex biological samples. More recently, we helped both enhance the detection of phenotype-defining transcripts using these methods and simplify their on-site processing for clinical applications. In parallel, we have also worked to democratize these techniques, providing open access to resources and protocols, training thousands locally and abroad, and establishing infrastructure and on-site collaborations spanning across 6 continents and 26+ countries.
Representative Publication: Shalek et al., Nature, 2013
Representative Publication: Gierahn et al., Nature Methods, 2017
Representative Publication: Hughes et al., Immunity, 2020
 Alex K. Shalek
Alex K. Shalek
 Andrew Navia
Andrew Navia
 Brittany Goods
Brittany Goods
 Carly Ziegler
Carly Ziegler
 Jay Prakadan
Jay Prakadan
 José Ordovas-Montañes
José Ordovas-Montañes
 Marc Wadsworth II
Marc Wadsworth II
 Sam Kazer
Sam Kazer
 Travis Hughes
Travis Hughes
 Vincent Miao
Vincent Miao
 Biology
Biology
 Cancer
Cancer
 Cell Atlas
Cell Atlas
 Chemistry
Chemistry
 Computational Methods
Computational Methods
 Genomics
Genomics
 Immunology
Immunology
 Infectious Disease
Infectious Disease
 Physics
Physics
 R&D
R&D
 Statistics
Statistics
 Technology
Technology
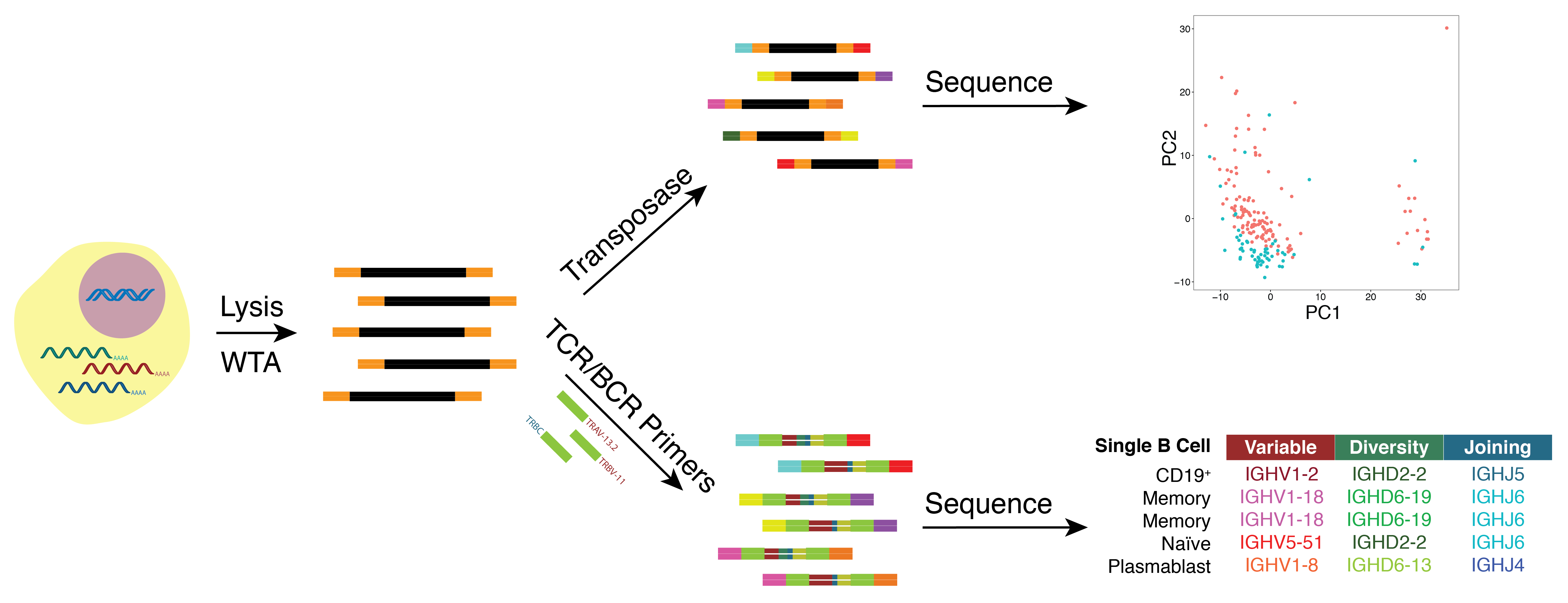
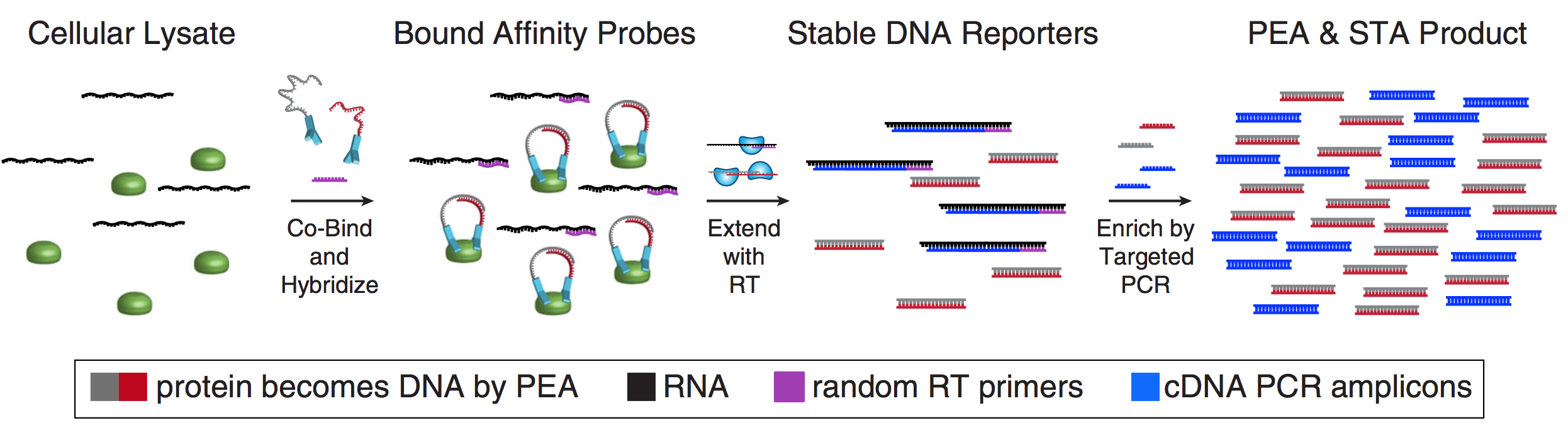
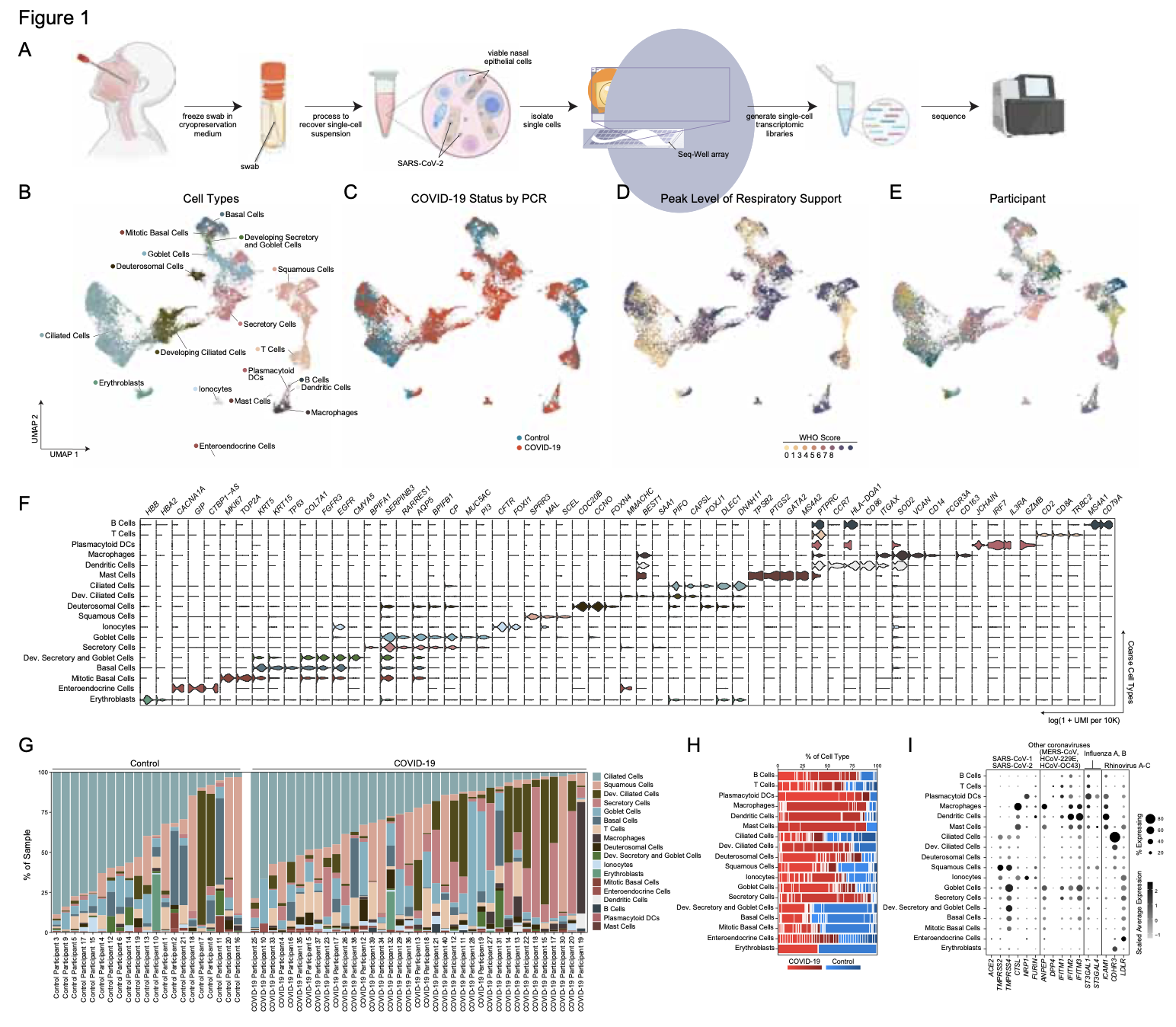
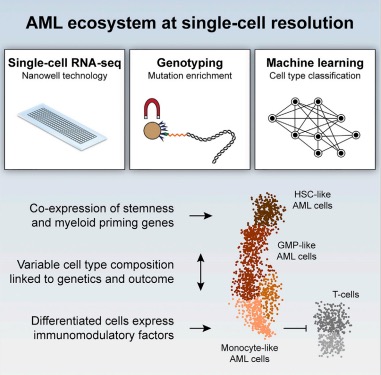
As many factors define cellular phenotype and influence disease beyond mRNA, we develop complementary methods for co-assaying other cellular attributes to enrich our understanding of the drivers of cellular behaviors. Examples including the abundance of additional ‘-omes’, the sequence and amount of important transcripts, cellular history, biophysical properties, spatial position, and functional output. Recently, we have worked to: 1. detect pathogens in cells and potentially actionable associated host factors; 2. query for specific mutations to identify cancer cells; and, 3. extract T cell receptor sequences to examine clonality. We have also formulated computational methods to derive deeper insights from these data (e.g., to examine viral dynamic in infected cells, reproducible features hidden by inter-individual variability, multicellular immune dynamics, intercellular communication, or alteration in cellular ecosystems associated with pathology).
Representative Publication: Tu et al., Nature Immunology, 2019
Representative Publication: Ziegler et al., Cell, 2021
Representative Publication: van Galen et al., Cell, 2019
 Aleth Gaillard
Aleth Gaillard
 Alex Genshaft
Alex K. Shalek
Carly Ziegler
Jay Prakadan
Marc Wadsworth II
Alex Genshaft
Alex K. Shalek
Carly Ziegler
Jay Prakadan
Marc Wadsworth II
 Sam Allon
Sam Kazer
Travis Hughes
Vincent Miao
Biology
Cancer
Chemistry
Computational Methods
Genomics
Immunology
Infectious Disease
Sam Allon
Sam Kazer
Travis Hughes
Vincent Miao
Biology
Cancer
Chemistry
Computational Methods
Genomics
Immunology
Infectious Disease
 Microbiology
Physics
R&D
Statistics
Technology
Microbiology
Physics
R&D
Statistics
Technology
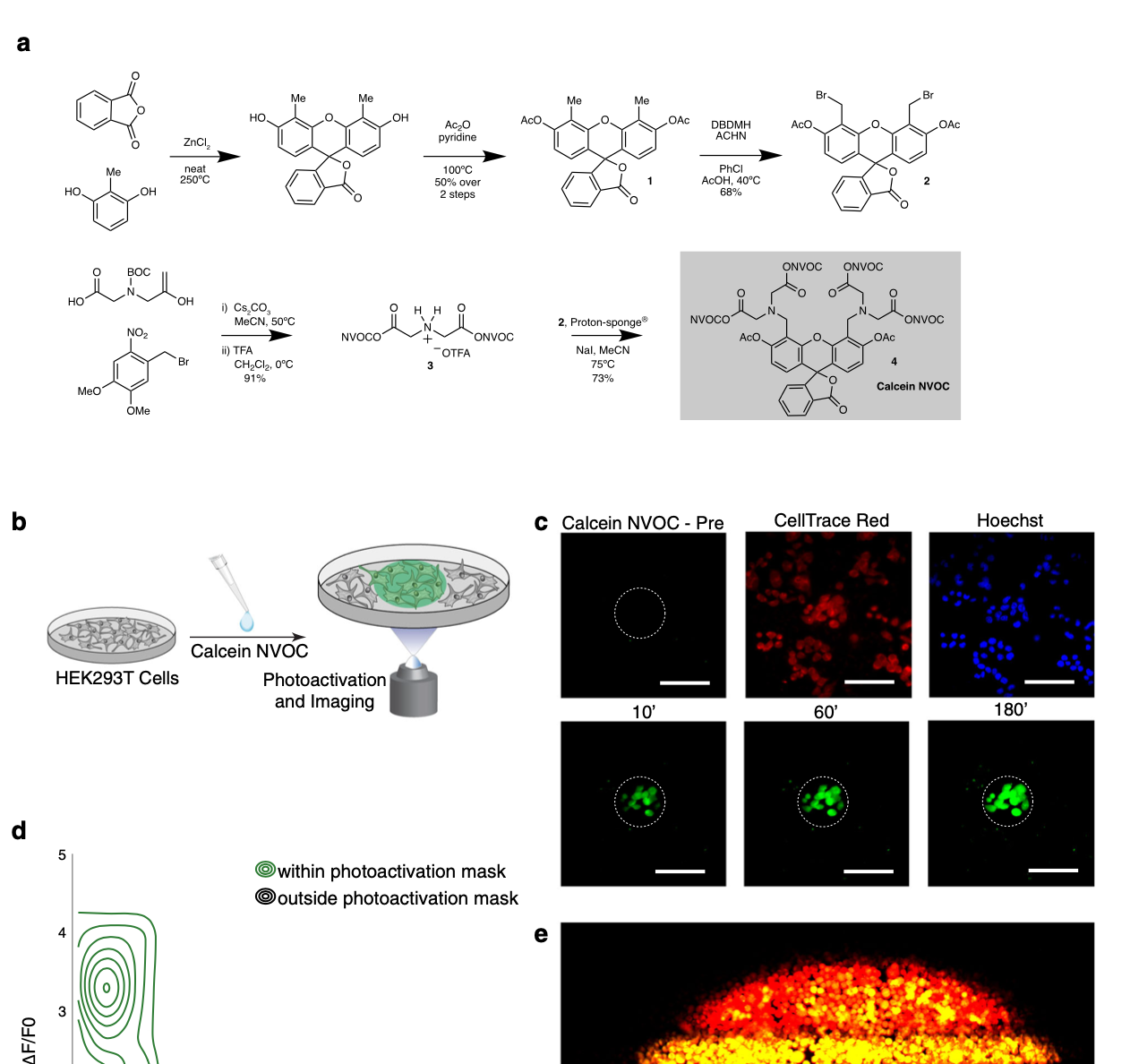
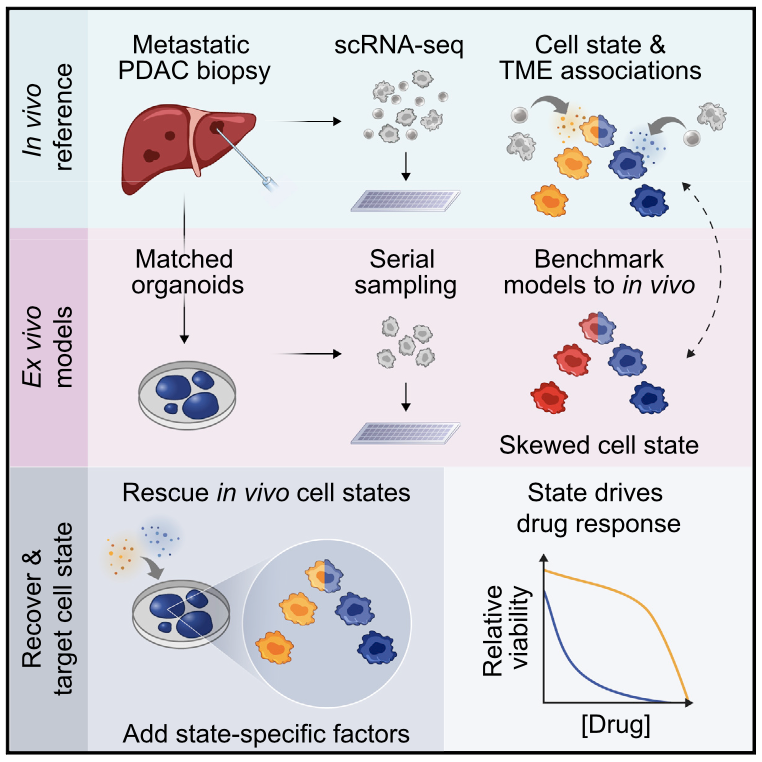
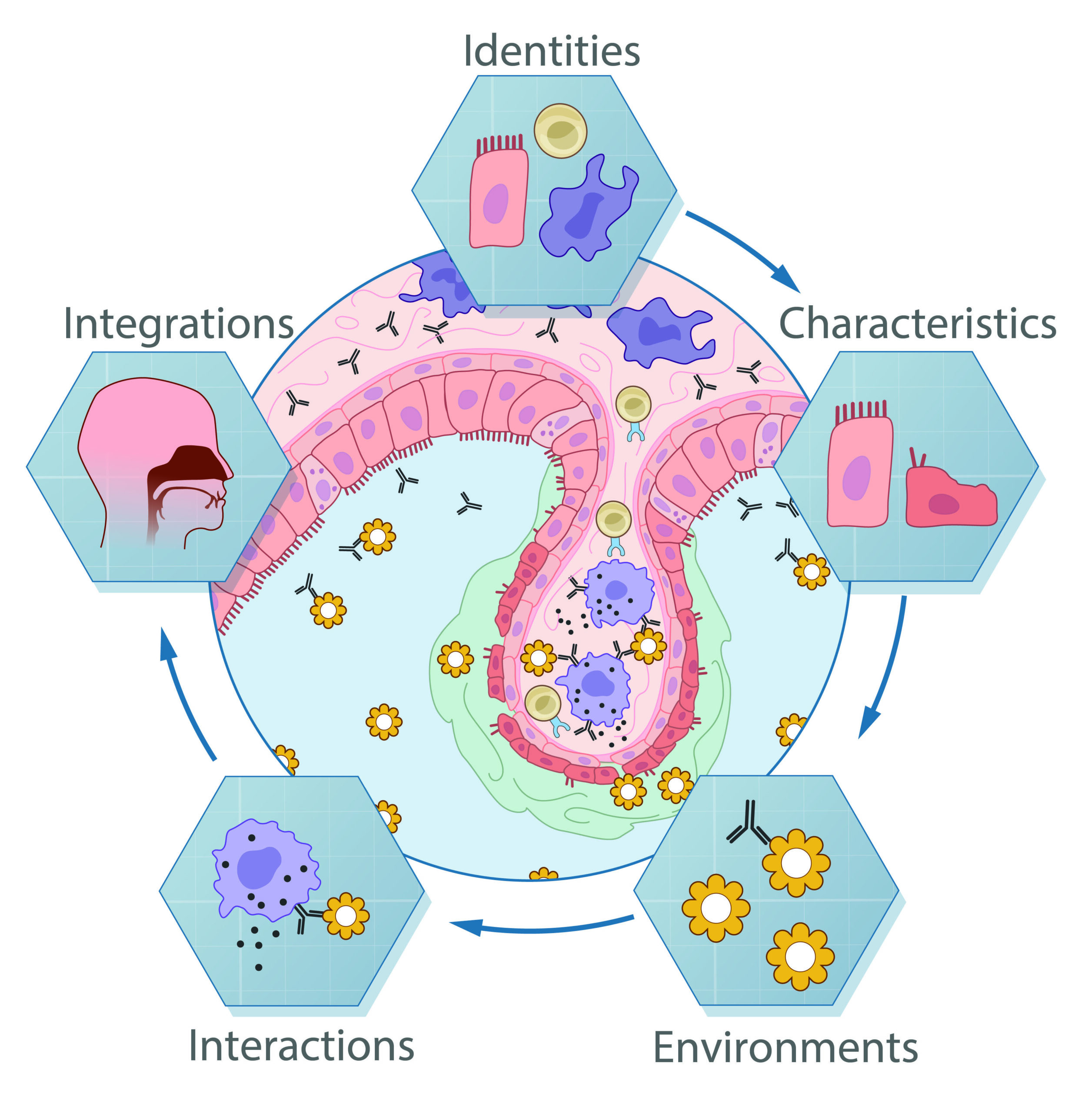
We explore how the extracellular milieu influences cellular decision-making. Here, we have employed controlled culture conditions with cells and organoids, chemical and genetic perturbations, and constant microfluidic perfusion. We also have leveraged natural microenvironmental variation within and across tissues via microdissection and by using photoactivatable probes that retain spatial information through dissociation. In each instance, we aim to understand the degree to which extracellular environments modulate, and can be used to rationally control, the responses of individual cells or the overall distribution thereof, with an eye toward engineering tissue responses.
Representative Publication: Genshaft et al., Nature Communications, 2021
Representative Publication: Mead et al., Nature Biomedical Engineering, 2022
Representative Publication: Raghavan et al., Cell, 2021
 Ben Mead
Carly Ziegler
Ben Mead
Carly Ziegler
 Constantine Tzouanas
Constantine Tzouanas
 Jennyfer Galvez-Reyes
Jennyfer Galvez-Reyes
 Josh Bromley
Josh Bromley
 Nolawit Mulugeta
Nolawit Mulugeta
 Peter Winter
Peter Winter
 Riley Drake
Sam Allon
Vincent Miao
Biology
Cancer
Chemistry
Computational Methods
Immunology
Infectious Disease
Microbiology
Physics
R&D
Statistics
Technology
Riley Drake
Sam Allon
Vincent Miao
Biology
Cancer
Chemistry
Computational Methods
Immunology
Infectious Disease
Microbiology
Physics
R&D
Statistics
Technology
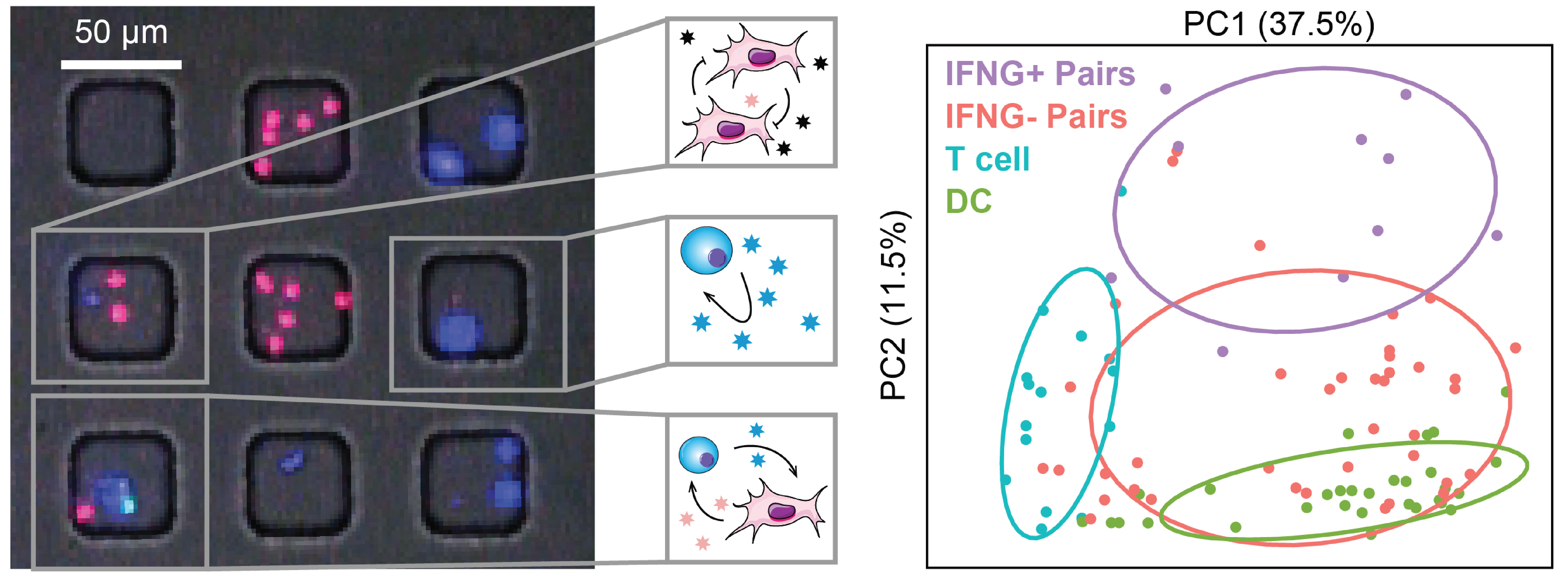
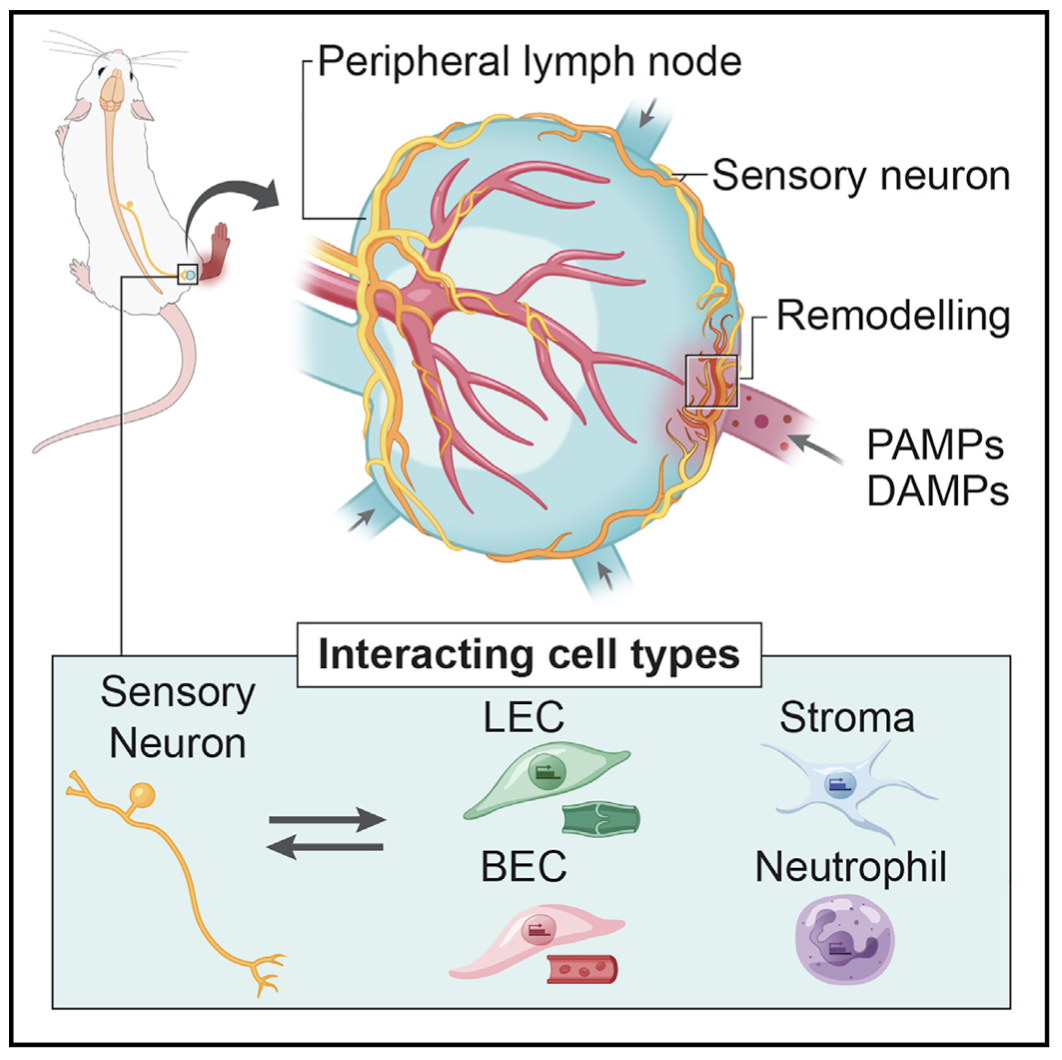
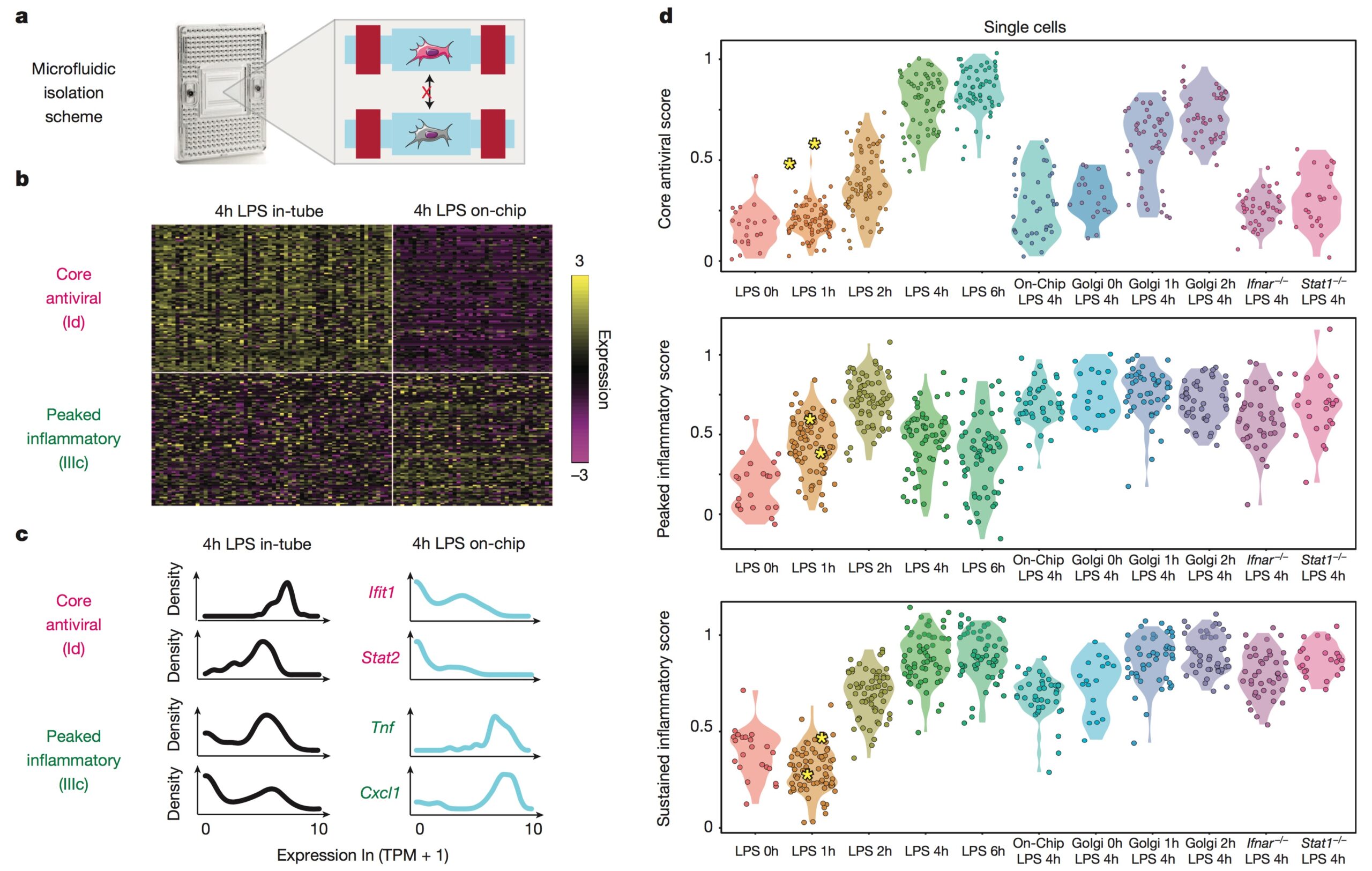
We examine the impact of intercellular interactions on cellular function. We have used coculture, imaging and perturbation strategies, as well as matched computational methods, to reinforce findings from dissociated samples, validate inferred cell-cell communication in vivo (e.g., between sensory neurons and lymph node resident cells), and manipulate multicellular systems (e.g., organoids). We are currently working on building arrayed, synthetically-designed cellular ensembles to examine how ‘tissue’ structure impacts functional response. Our overall goal is to understand cellular co-dependencies that influence niche- and tissue-level response dynamics.
Representative Publication: Huang et al., Cell, 2020
Representative Publication: Prakadan et al., Nature Communications, 2021
Representative Publication: Martin-Gayo et al., Genome Biology, 2018
 Alex Genshaft
Andrew Navia
Brittany Goods
Carly Ziegler
Jay Prakadan
José Ordovas-Montañes
Alex Genshaft
Andrew Navia
Brittany Goods
Carly Ziegler
Jay Prakadan
José Ordovas-Montañes
 Kellie Kolb
Marc Wadsworth II
Kellie Kolb
Marc Wadsworth II
 Marko Vukovic
Marko Vukovic
 Sarah Nyquist
Biology
Cancer
Chemistry
Computational Methods
Genomics
Immunology
Infectious Disease
Microbiology
Physics
R&D
Statistics
Technology
Sarah Nyquist
Biology
Cancer
Chemistry
Computational Methods
Genomics
Immunology
Infectious Disease
Microbiology
Physics
R&D
Statistics
Technology
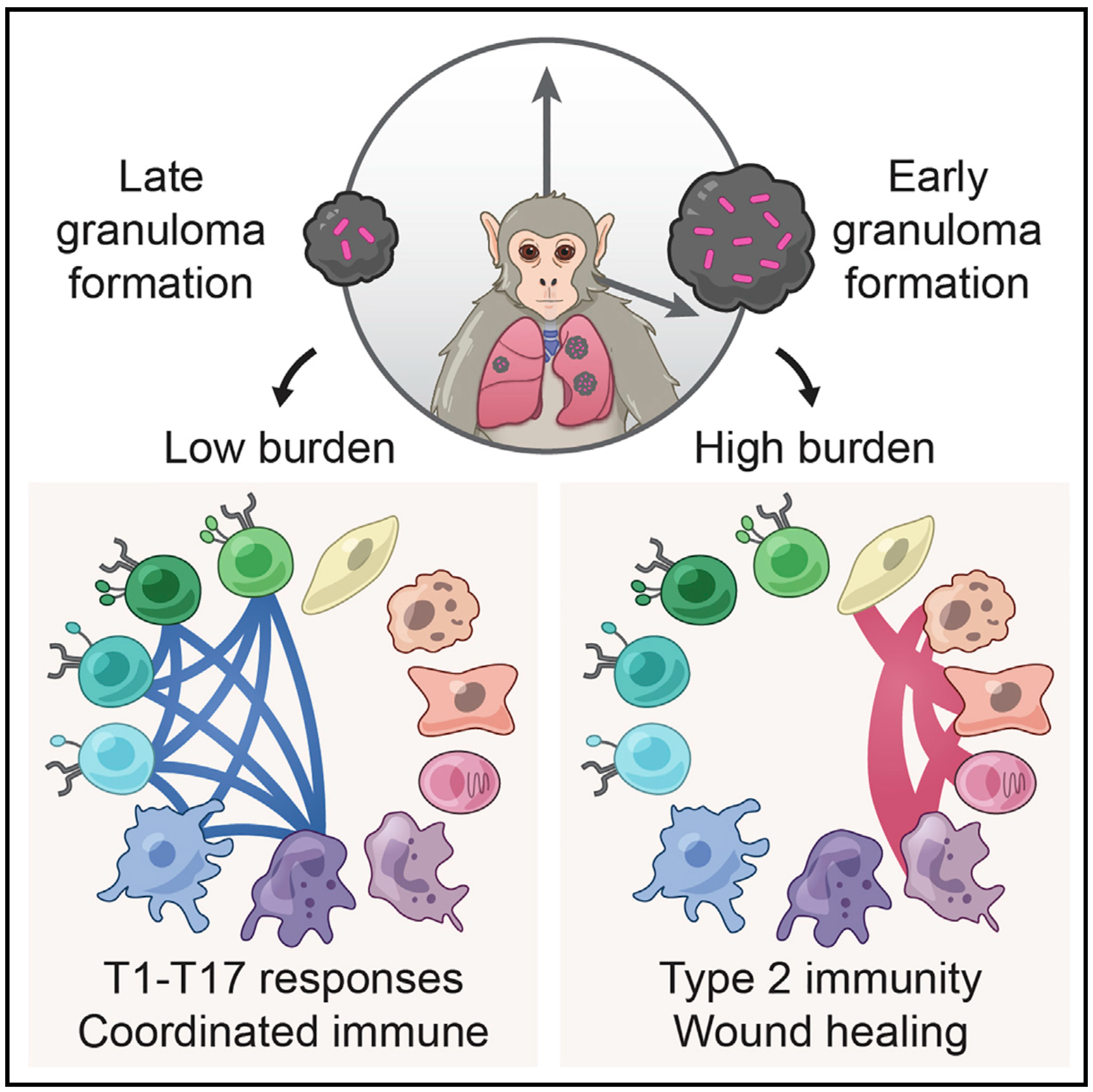
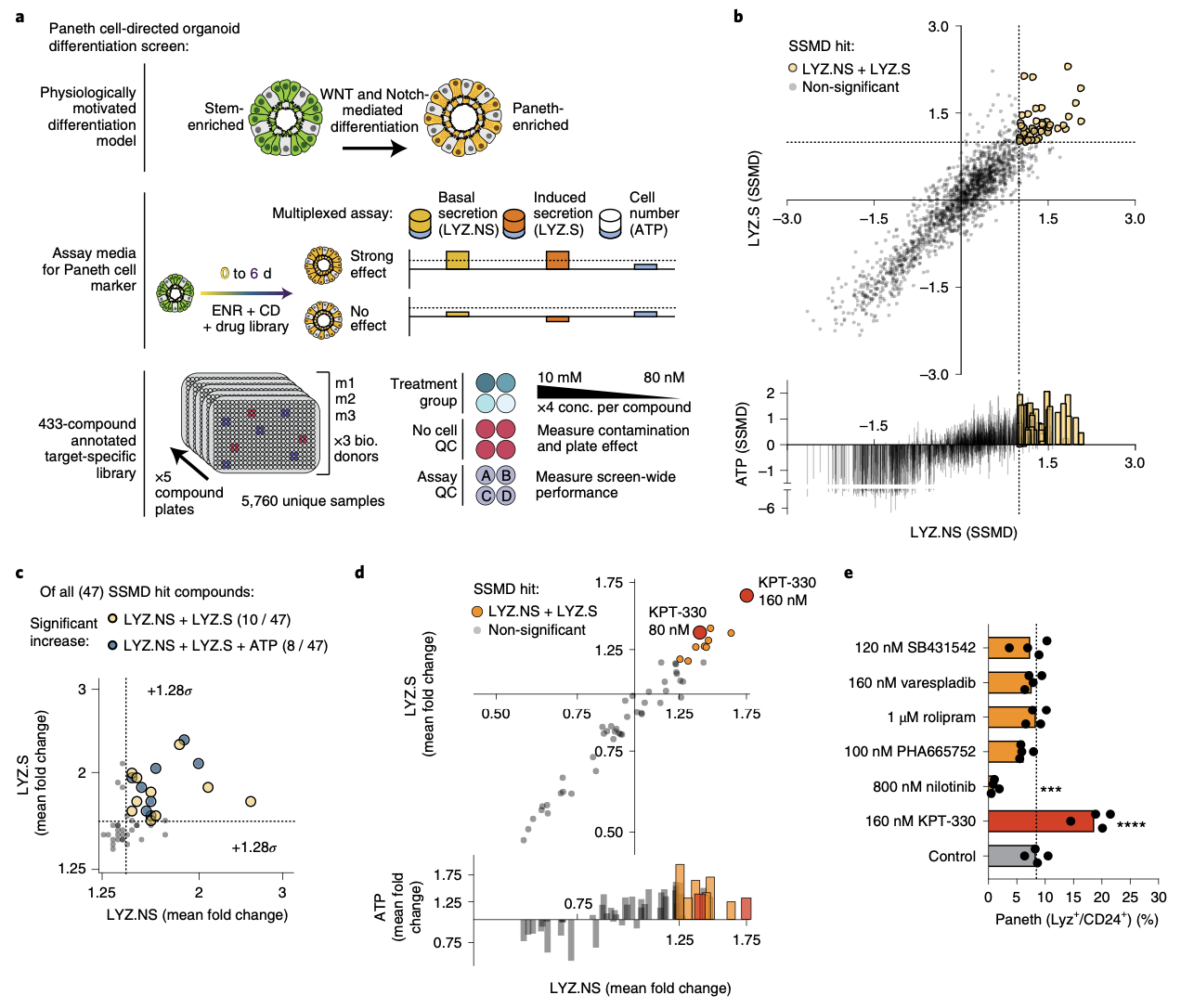
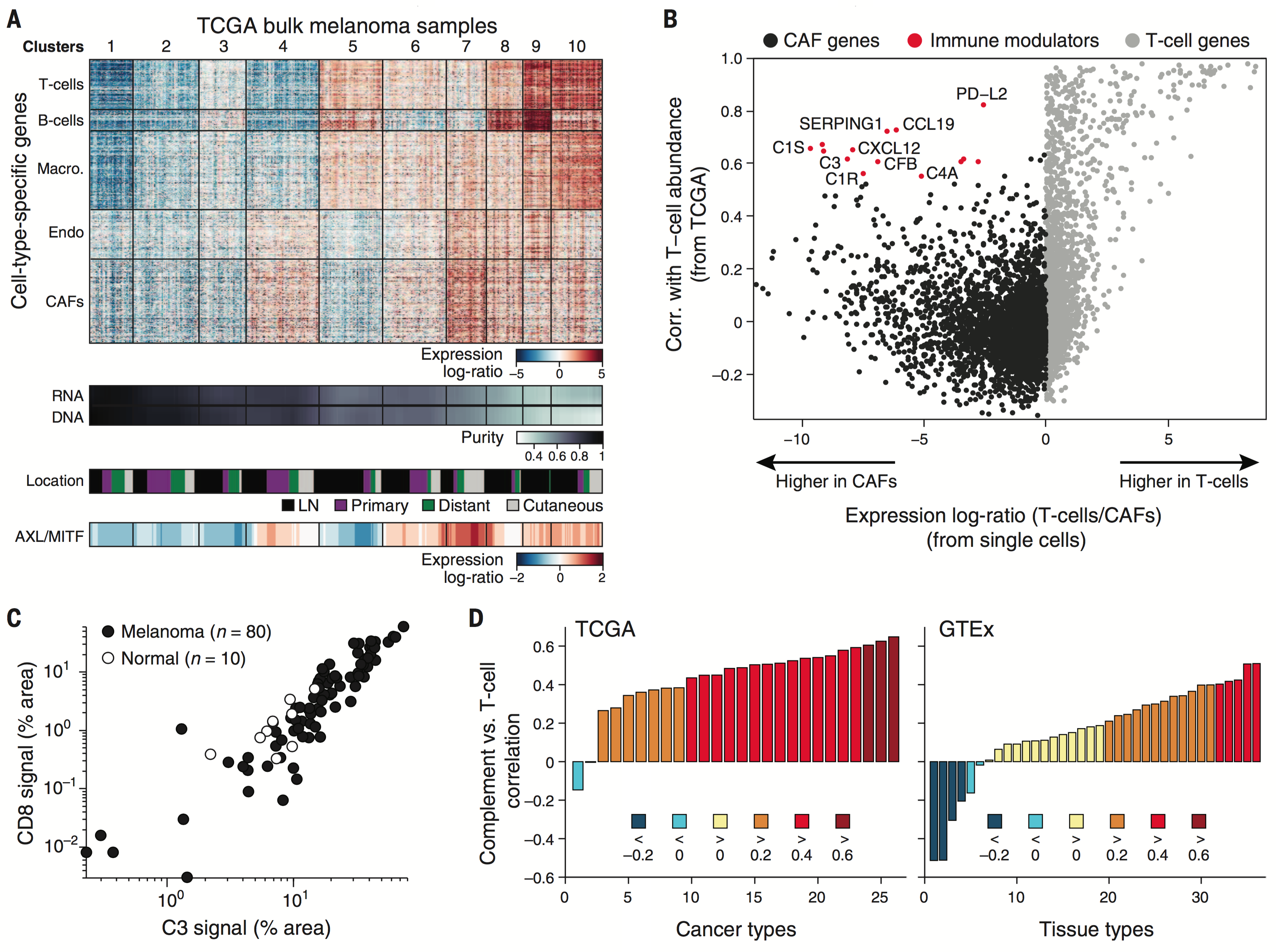
We broadly study how intra- and extracellular circuits collectively drive healthy and diseased tissue states. By leveraging the massive genomic datasets we and others have generated from complex tissues (like melanoma tumors, inflamed gut, and nasal polyps), we have begun to identify common and unique cell types/states and circuits associated with pathology that may be important for regulating biological function and stability. Our current findings suggest multiple overlaps among distinct diseases, pointing to the possibility of a finite set of evolved response strategies and thus common interventions based on adjusting specific cell states, cell frequencies, and/or cell-cell communication pathways.
Representative Publication: Ziegler et al., Cell, 2020
Representative Publication: Gideon et al., Immunity, 2022
Representative Publication: Ordovas-Montanes et al., Nature, 2018
 Ben Mead
Ben Mead
 Benjamin Doran
Benjamin Doran
 Conner Kummerlowe
Constantine Tzouanas
Jay Prakadan
Marc Wadsworth II
Marko Vukovic
Conner Kummerlowe
Constantine Tzouanas
Jay Prakadan
Marc Wadsworth II
Marko Vukovic
 Samira Ibrahim
Sarah Nyquist
Vincent Miao
Biology
Cancer
Cell Atlas
Computational Methods
Genomics
Immunology
Samira Ibrahim
Sarah Nyquist
Vincent Miao
Biology
Cancer
Cell Atlas
Computational Methods
Genomics
Immunology
 Medicine
R&D
Statistics
Medicine
R&D
Statistics